¿Qué es el síndrome de Mayer-Rokitansky-Küster-Hauser?
El síndrome de Mayer-Rokitansky-Küster-Hauser se conoce por su sigla en inglés como MRKHS o, simplemente, como síndrome de Rokitansky.
Esta patología también se llama agenesia mülleriana o hipoplasia mülleriana, puesto que se debe a un defecto durante el desarrollo de los conductos de Müller que forman el útero, las trompas de Falopio y la vagina en la embriogénesis.
Las mujeres que sufren el síndrome de Rokitansky pueden tener problemas para mantener relaciones sexuales si no desarrollan la vagina, además del trauma que supone para algunas no poder tener hijos. Por tanto, será necesario adoptar algunas medidas para mejorar la calidad de vida de estas mujeres.
A continuación tienes un índice con todos los puntos que vamos a tratar en este artículo.
Índice
- 1.
- 2.
- 3.
- 4.
- 4.1.
- 4.2.
- 5.
- 6.
- 6.1.
- 6.2.
- 6.3.
- 6.4.
- 6.5.
- 7.
- 8.
Síndrome de MRKH y ausencia de útero
El síndrome de Rokitansky es un trastorno congénito del aparato reproductor femenino que afecta a 1 de cada 5.000 mujeres en el mundo. Estas mujeres ya nacen sin útero y sin trompas de Falopio. En cuanto a la vagina, según el grado de afección, pueden tener ausencia total o parte de ella.
La agenesia mülleriana implica la ausencia de vagina, útero y trompas en la mujer, pero con ovarios funcionales.
Las causas del síndrome de Rokitansky no se conocen en profundidad. Se debe a un fallo en el desarrollo embrionario temprano, sobre las semanas 4 y 12 de gestación. Los conductos de Müller del feto de sexo femenino no se desarrollan y, como consecuencia, el útero, las trompas y la vagina no se forman de la manera adecuada.

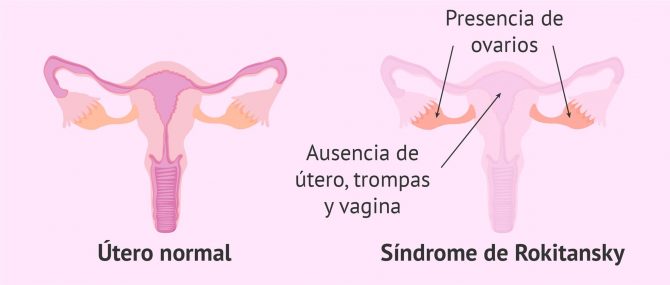
En cambio, los ovarios se desarrollan a partir de una estructura diferente a los conductos de Müller y, por tanto, tienen forma normal y son funcionales en las mujeres con síndrome de Rokitansky.
¿Cómo es el diagnóstico del síndrome de Rokitansky?
Una mujer con el síndrome de Rokitansky tiene producción normal de sus hormonas femeninas, por lo que llega a la pubertad y desarrolla sus características sexuales femeninas. Puesto que, como hemos dicho, presenta ovarios funcionales, la ovulación también es normal.
Sin embargo, la menstruación nunca llega con la entrada a la pubertad y, normalmente, las niñas acuden a su primera visita ginecológica por este motivo.
El síndrome de Rokitansky suele diagnosticarse a los 15-18 años mediante una ecografía o una resonancia magnética.
Con un examen ginecológico básico, no es posible diagnosticar esta patología, ya que los genitales externos son de apariencia normal.
Por otra parte, con la ecografía de ultrasonido, el ginecólogo puede comprobar si hay una ausencia completa o parcial de la vagina, el cuello uterino y/o la matriz. En ocasiones, es posible observar la presencia de un saco vaginal corto o un útero muy rudimentario si la ausencia no es total.
Por último, la resonancia magnética nuclear confirmará estos resultados.
Síntomas y consecuencias
Los síntomas que presenta una mujer con el síndrome de Rokitansky van a depender del grado de afectación.
En general, la amenorrea primaria (ausencia de menstruación) es el primer síntoma que detectan estas mujeres con la llegada a la pubertad.
Otras consecuencias o síntomas de la agenesia mülleriana se detallan a continuación:
- Relaciones sexuales difíciles y dolorosas en mujeres con la vagina acortada.
- Incapacidad de penetración vaginal cuando la vagina no está formada.
- Enfermedades óseas a nivel vertebral en el 12-50% de los casos, aunque es posible que también haya un efecto sobre las extremidades.
- Malformaciones renales como el riñón pélvico, el riñón en herradura, la hidroureteronefrosis y la duplicidad ureteral, aunque de forma menos común.
- Infertilidad femenina por la imposibilidad de gestar al no tener útero.


Como hemos dicho en el anterior apartado, la mujer sí tiene ovarios funcionales y producción de hormonas sexuales. Por tanto, sí que tiene un desarrollo sexual adecuado, con la aparición de los caracteres sexuales secundarios: crecimiento de los pechos, aparición de vello, etc.
Tratamiento
El síndrome de Rokitansky no tiene cura en la actualidad. No obstante, existen técnicas enfocadas a tratar la aplasia vaginal (ausencia de vagina) con el objetivo de permitir a estas mujeres llevar una vida sexual activa y un estilo de vida lo más normal posible.
A continuación, vamos a dividir los tipos de tratamiento en función de si es necesario recurrir a la cirugía o no. En ambos casos, es necesario que la mujer adolescente haya llegado a la madurez sexual y emocional para poder aplicar estas técnicas.
Técnicas no quirúrgicas
Este tipo de tratamiento sin cirugía consiste en dilataciones vaginales y, por ello, será necesario que la mujer posea vagina aunque sea pequeña. Por ejemplo, una vagina de 4 cm se podrá convertir de una de 8-10 cm con lubricación y buena distensibilidad.
El dilatador de Frank es el método más conocido para crear una vagina óptima sin recurrir a la cirugía. Este método consiste en unos tubos de plástico de diferente tamaño que ayudan a aumentar la vagina gradualmente con la aplicación de una presión intermitente.

Su duración aproximada es de seis semanas a varios meses y su efectividad es muy elevada. En caso de no tener éxito o no ser posible el uso de dilatadores, será necesario recurrir a una intervención quirúrgica para crear una neovagina.
Técnicas quirúrgicas
Son las más apropiadas cuando la mujer tiene una ausencia total de vagina o cuando las dilataciones no han sido efectivas. Estas técnicas consisten en construir un canal similar a la vagina entre el espacio recto-vesical.
La más utilizada en este caso es la técnica de McIndoe. Consiste en utilizar un injerto de piel de la paciente y colocarlo sobre una prótesis con forma de pene de silicona.
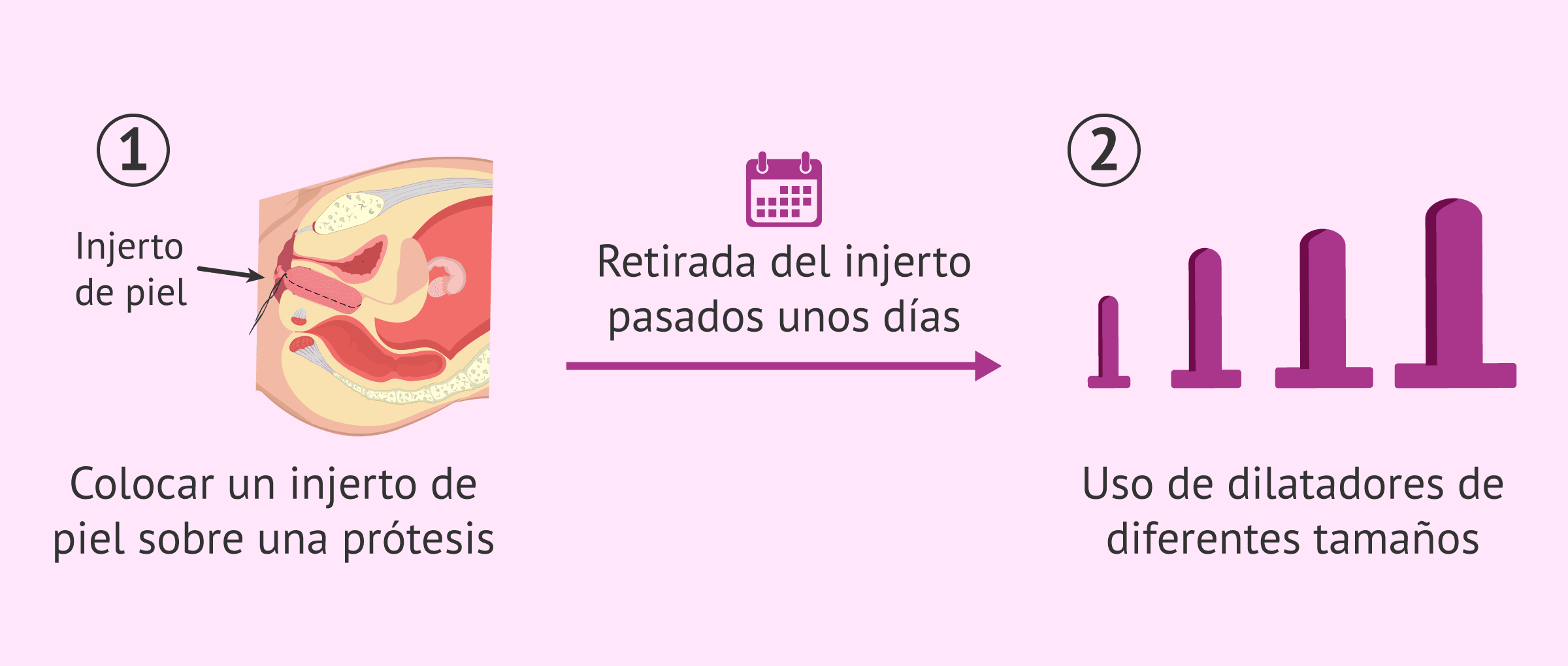
Esta prótesis con el injerto se amolda y se fija al túnel vaginal con sutura. Después de unos días, se retira la prótesis y se utilizan dilatadores durante varias semanas.
Además de estos tratamientos descritos aquí, el apoyo psicológico será indispensable en estas pacientes, pues sufren mucho al descubrir que padecen este transtorno en su sistema reproductor.
Síndrome de Rokitansky y embarazo
Las mujeres que no tienen útero no pueden quedar embarazadas de manera natural ni gestar un bebé. No obstante, tienen óvulos viables en sus ovarios y, por tanto, pueden tener descendencia biológica gracias a técnicas de reproducción asistida, como la fecundación in vitro (FIV) de sus óvulos en el laboratorio.
Por otra parte, es indispensable encontrar una solución para que la gestación del bebé durante los 9 meses sea posible. Las dos opciones para poder llevar a cabo un embarazo y el nacimiento del un niño sano son las siguientes:
- Trasplante de útero
- es una técnica experimental que no se utiliza de manera rutinaria. En 2014, nació el primer bebé de un útero trasplantado en Suecia.
- Gestación subrogada
- los óvulos fecundados de la mujer sin útero son transferidos a una mujer que se ofrece como gestante para mantener el embarazo y dar a luz al bebé de la otra mujer o pareja. Esta técnica reproductiva no es legal en España en la actualidad. Por tanto, los padres que necesiten recurrir a ella tendrán que viajar a países como EE.UU., Canadá y Ucrania, entre otros.
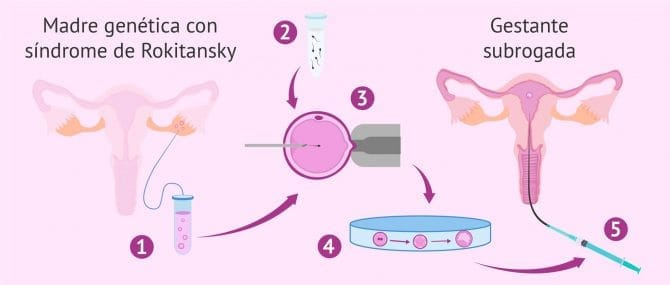
Si necesitas obtener más información sobre la técnica de gestación subrogada, conocida vulgarmente como vientre de alquiler, te recomendamos visitar el siguiente post: ¿Qué es la gestación subrogada?
Preguntas de los usuarios
¿Cómo se diagnostica el síndrome de Rokitansky?
El síndrome de Rokitansky-Kuster-Hausser es una malformación del aparato genital femenino que se caracteriza por la ausencia de parte del trato genital. Por lo tanto se las mujeres que presentan este síndrome presentan vulva y entrada vaginal, pero la ausencia de la parte superior de la vagina y el útero.
Debido a unos órganos reproductores externos aparentemente normales, es fácil comprender que la mayoría de pacientes no sean diagnosticadas durante la infancia.
Cuando llega la pubertad, la ausencia de amenorrea primaria ( la ausencia de menstruación) puede poner en alerta al doctor que atienda a la paciente. Otra situación común que oriente al médico al estudio de este síndrome es en pacientes que reportan problemas en sus primeras relaciones sexuales.
Una vez el médico intuye que la paciente puede padecer este síndrome el diagnóstico es sencillo: la exploración vaginal que determine que la vagina es muy corta y una ecografía que demuestra la ausencia de los dos tercios superiores de útero y vagina.
Leer más
¿Por qué sangro si no tengo útero ni ovarios?
Las mujeres con el síndrome de Rokitansky pueden presentar pequeños sangrados y dolor pélvico por la existencia de una pequeña parte de endometrio funcional en un útero rudimentario, incluso por la formación de miomas en este mismo.
Sin embargo, lo más común es que estas mujeres tengan una ausencia total de útero. En este caso, tendrán que hacerse las pruebas necesarias para averiguar de dónde viene el sangrado.
¿Qué cariotipo tienen las mujeres con el síndrome de Rokitansky?
El síndrome de agenesia mülleriana, también conocido como síndrome de Rokitansky, está causado por un defecto en el desarrollo fetal. Sin embargo, este síndrome no está provocado por ninguna alteración genética, por lo que no hay alteraciones en el cariotipo.
Por tanto, las niñas que nacen sin útero, tienen un cariotipo normal (46,XX), es decir, poseen 46 cromosomas y los cromosomas sexuales son XX.
¿Puedo quedar embarazada si no tengo útero?
Evidentemente, no. El útero es el órgano esencial para lograr un embarazo. Una mujer con el síndrome de Rokitansky, es decir, sin útero no puede mantener un embarazo. Estas mujeres tienen tres alternativas si desean ser mamás:
- La adopción.
- La gestación subrogada, aunque en España este tratamiento no es legal.
- El trasplante de útero (en etapa experimental).
Si no tengo útero, ¿puedo tener cáncer de cuello uterino?
No, no es posible. Evidentemente, no se puede desarrollar un cáncer en un órgano que no existe.
Esto no quiere decir que la mujer no corra el riesgo de sufrir un tumor cancerígeno en otros órganos del sistema reproductor, como son los ovarios, la vagina o las trompas de Falopio.
Lectura recomendada
Para que una mujer sea fértil, debe tener un desarrollo adecuado de todo su sistema reproductor, tanto interno como externo. Para obtener más información sobre este tema, te recomendamos leer el siguiente post: La fertilidad en la mujer.
La agenesia mülleriana es la más severa de las malformaciones uterinas congénitas. No obstante, existen otras anomalías que implican un problema de infertilidad. Si quieres leer más sobre esto, puedes entrar en el siguiente artículo: Malformaciones uterinas en la mujer.
Hacemos un gran esfuerzo para ofrecerte información de máxima calidad.
🙏 Por favor, comparte este artículo si te ha gustado. 💜💜 ¡Nos ayudas a seguir!
Bibliografía
John C Petrozza. Mayer-Rokitansky-Küster-Hauser syndrome and associated malformations: are they as common as we think? Fertil Steril. 2016 Oct;106(5):1047-1048. doi: 10.1016/j.fertnstert.2016.06.033 (Ver)
Karine Morcel, Laure Camborieux; Programme de Recherches sur les Aplasies Müllériennes; Daniel Guerrier. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007 Mar 14:2:13. doi: 10.1186/1750-1172-2-13 (Ver)
Laura Londra, Farah S Chuong, Lisa Kolp. Mayer-Rokitansky-Kuster-Hauser syndrome: a review. Int J Womens Health. 2015 Nov 2:7:865-70. doi: 10.2147/IJWH.S75637 (Ver)
Magdalena Liszewska-Kapłon, Mateusz Strózik, Łukasz Kotarski, Maciej Bagłaj, Lidia Hirnle. Mayer-Rokitansky-Küster-Hauser syndrome as an interdisciplinary problem. Adv Clin Exp Med. 2020 Apr;29(4):505-511. doi: 10.17219/acem/118850 (Ver)
Morten Krogh Herlin, Michael Bjørn Petersen, Mats Brännström. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. 2020 Aug 20;15(1):214. doi: 10.1186/s13023-020-01491-9 (Ver)
Susanne Ledig, Peter Wieacker. Clinical and genetic aspects of Mayer-Rokitansky-Küster-Hauser syndrome. Med Genet. 2018;30(1):3-11. doi: 10.1007/s11825-018-0173-7. Epub 2018 Feb 21 (Ver)