¿Qué es el Síndrome de Kallmann? – Hipogonadismo en hombres y mujeres
El Síndrome de Kallmann, también conocido como Síndrome de Maestre-Kallmann-Morsier, es una enfermedad genética asociada con el hipogonadismo hipogonadotrópico y los trastornos del sentido del olfato, como la anosmia y la hiposmia.
Esta patología puede aparecer tanto en hombres como en mujeres, aunque es más común que aparezca en los primeros. La prevalencia del síndrome de Kallmann es de 1 en cada 8.000 varones y de 1 en cada 40.000 mujeres.
Puesto que esta enfermedad afecta a todo el eje hipotálamo-hipófisis y a la producción de las hormonas sexuales, el síndrome de Kallmann provoca infertilidad.
A continuación tienes un índice con todos los puntos que vamos a tratar en este artículo.
Índice
- 1.
- 2.
- 2.1.
- 2.2.
- 2.3.
- 3.
- 4.
- 5.
- 5.1.
- 5.2.
- 5.3.
- 5.4.
- 6.
- 7.
Definición y causas
El Síndrome de Kallmann (SK), también escrito Kallman, o Síndrome de Maestre-Kallmann-Morsier, es una enfermedad hereditaria que debe su nombre al psiquiatra de origen alemán Franz Josef Kallmann (1897-1965).
Este síndrome tiene un origen genético y, en función del tipo de mutación, su herencia será autosómica dominante, autosómica recesiva o ligada al cromosoma X.
La principal causa de esta enfermedad es la deficiencia de la hormona GnRH (hormona liberadora de gonadotropinas) debido a un fallo durante el desarrollo embrionario del individuo.
Concretamente, se produce un defecto en la migración de las neuronas hipotalámicas que sintetizan la GnRH desde el epitelio olfatorio hasta la región hipotalámica del cerebro.
Además, también se produce un defecto en el desarrollo del sistema olfatorio y, por ello, las personas con síndrome de Kallmann sufren de anosmia o hiposmia.


La GnRH es una de las hormonas que controlan el eje hipotálamo-hipofisiario. La liberación de la GnRH por el hipotálamo estimula la secreción de las gonadotropinas FSH y LH por parte de la hipófisis.
A su vez, estas hormonas hipofisiarias son las encargadas de regular todo el desarrollo sexual en hombres y en mujeres.
¿Qué síntomas causa el síndrome de Kallmann?
Debido al hipogonadismo hipogonadotropo que provoca el síndrome de Kallmann, los afectados tendrán una maduración incompleta del sistema reproductor y una falta de desarrollo sexual.
Como consecuencia, tanto hombres como mujeres tendrán unos caracteres sexuales secundarios mal definidos una vez lleguen a la pubertad, además de problemas de esterilidad para tener hijos en el futuro.
A continuación, vamos a comentar las manifestaciones más importantes del síndrome de Kallmann en ambos sexos:
En los hombres
En función de la edad y la etapa del desarrollo del varón, será posible detectar distintas alteraciones relacionadas con el síndrome de Kallmann.
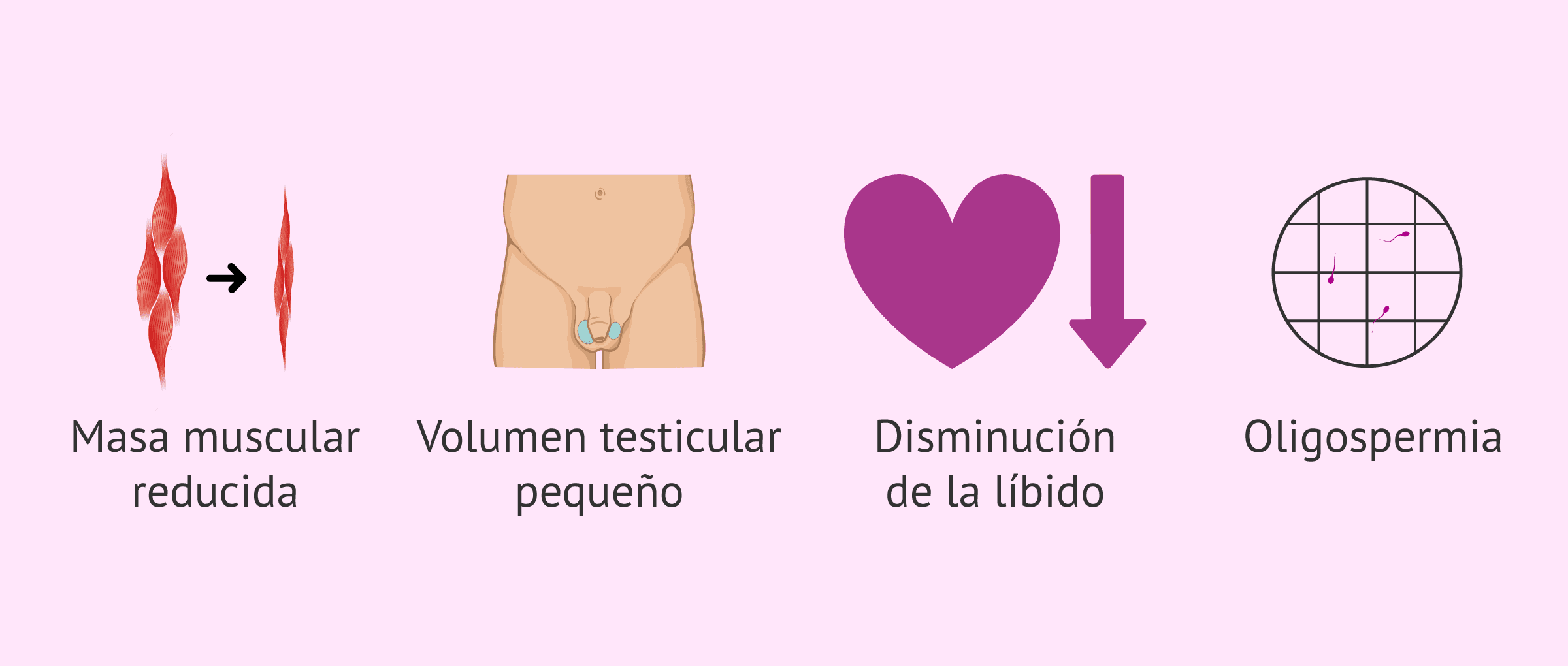
Después de la pubertad y una vez llegados a la edad adulta, los hombres con síndrome de Kallmann tendrán un fenotipo relacionado con el hipogonadismo y la falta de la hormona testosterona:
- Densidad ósea y masa muscular reducida
- Volumen testicular pequeño
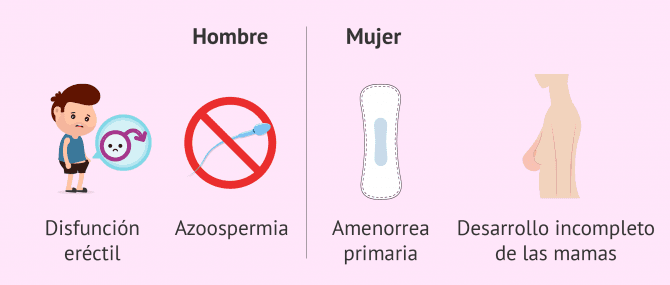
- Disfunción eréctil
- Disminución de la libido
- Oligospermia o azoospermia

Estos individuos también suelen tener una apariencia de adolescente, ya que sus caracteres infantiles persisten en la vida adulta.
En las mujeres
Los síntomas principales del síndrome de Kallmann en las mujeres son la amenorrea primaria y el desarrollo incompleto o ausente de las mamas al llegar a la pubertad.
Debido a la falta de las hormonas sexuales femeninas, como los estrógenos, estas mujeres serán anovuladoras y, por tanto, también sufrirán esterilidad.
Otros síntomas
Uno de los síntomas más característicos del síndrome de Kallmann en ambos sexos, como ya hemos comentado, es la ausencia parcial o completa de la percepción de los olores debido al desarrollo incorrecto de los bulbos olfativos.
Por otra parte, estas personas también pueden presentar de forma menos frecuente las siguientes alteraciones:
- Anomalías renales, como la agenesia renal unilateral
- Retraso mental moderado
- Déficit auditivo
- Movimiento involuntario de las manos o de los globos oculares
- Paladar hendido
- Labio leporino

¿Cómo se diagnostica el síndrome de Kallmann?
Para determinar si una persona sufre realmente el síndrome de Kallmann, existen diferentes métodos diagnósticos:
- Análisis hormonal
- se miden las hormonas FSH y LH, así como las hormonas sexuales testosterona y estrógenos.
- Evaluación del olfato
- se valora cualitativa y cuantitativamente la percepción olfativa. También puede hacerse una resonancia magnética para ver los bulbos olfatorios.
- Evidencia clínica de hipogonadismo
- ausencia de caracteres secundarios, como la amenorrea, la disfunción eréctil, la falta de libido, etc.
- Test genético
- para identificar la mutación exacta que causa la enfermedad.

Antes de realizar el diagnóstico, es importante diferenciar bien entre un posible síndrome de Kallmann y un retraso normal del crecimiento y de la pubertad. Para ello, las pruebas diagnósticas deberán hacerse una vez pasada la pubertad, después de los 15-16 años.
Tratamiento del síndrome de Kallmann
El tratamiento del síndrome de Kallmann se encuentra enfocado a conseguir la virilización o feminización puberal de los pacientes.
Para ello, habrá que seguir una terapia hormonal sustitutiva que consiga desarrollar y mantener los caracteres sexuales secundarios. Por una parte, el vello corporal, la voz más grave, el desarrollo muscular y óseo en los hombres y, por otra parte, los pechos desarrollados, la menstruación, el vello púbico, etc. en las mujeres.
Por lo general, el tratamiento para el síndrome de Kallmann tiene que ser mantenido de por vida.
En el momento que una persona afectada por el síndrome de Kallmann desee tener hijos, habrá que cambiar el tratamiento hormonal para poder restaurar la fertilidad. A continuación, vamos a especificar algunos tratamientos en función del sexo:
- Varón con síndrome de Kallmann
- las inyecciones repetidas de GnRH promoverán la espermatogénesis y habrá espermatozoides a partir del tercer mes. También es posible probar con la administración de hCG seguida de FSH, ya que aumentarán la concentración de testosterona intratesticular.
- Mujer con síndrome de Kallmann
- se administrará medicación hormonal para provocar una estimulación ovárica suave y, a continuación, se programarán las relaciones sexuales. Esto es lo que se conoce como coito dirigido.
En caso de no conseguir el embarazo con estos tratamientos o si existen otras alteraciones más graves de la fertilidad, la pareja tendrá que recurrir a técnicas como la inseminación artificial (IA) o la fecundación in vitro (FIV).
También será interesante plantear el diagnóstico genético preimplantacional (DGP) con el objetivo de evitar la transmisión de esta enfermedad a la descendencia.
Es posible calcular TU probabilidad de embarazo según el tratamiento, edad y otros factores?
Nos parece demasiado importante como para no compartirlo contigo.
Puedes personalizar tu informe de fertilidad en 2 minutos.
Por último, en cuanto a la recuperación del sentido olfativo, no existe ningún tratamiento actualmente.
Preguntas de los usuarios
¿Puedo ser mamá si tengo hipogonadismo hipergonadotropo?
Las causas del hipogonadismo hipergonadotropo son múltiples y dan lugar a una alteración en la función ovárica, condicionando esto la fertilidad.
En términos generales podemos afirmar que la posibilidad de gestación se va a determinar según exista o no dotación folicular ovárica y a la integridad del factor uterino. En el caso de que se cumplieran ambos criterios sí sería posible la gestación mediante gestación espontánea o técnicas de Reproducción Asistida (inseminación artificial, fecundación in vitro o donación de gametos o embriones).
En cambio, si no existiera dotación folicular y se mantuviera integridad uterina sería posible la gestación mediante donación de gametos o embriones.
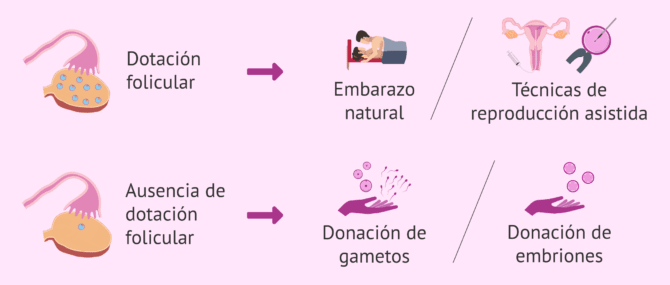
Es importante subrayar que en aquellos casos en los que el hipogonadismo hipergonadotropo se asocie a un síndrome genético sería recomendable realizar Diagnóstico Genético Preimplantacional en casos de fecundación in vitro.
¿El síndrome de Kallman tiene cura?
La causa del síndrome de Kallman es genética, con diferentes genes implicados. Por ello, no existe una cura como tal. El tratamiento consiste en dar de forma exógena las hormonas que necesita el cuerpo para que se produzca un desarrollo puberal correcto. En las mujeres se administran estrógenos y en los hombres testosterona, en ambos casos se mantendrán de forma indefinida.
Además, si llega un momento en el que el paciente desea tener hijos, se debe activar el eje hipotálamo hipofisario gonadal con medicación. Para ello puede administrarse GnRH o FSH y LH, para que activen ovarios y testículos.
Leer más
¿Cuáles son las características del síndrome de Kallman?
El síndrome de Kallman, también llamado Síndrome de Maestre-Kallmann.Morsier, consiste en un hipogonadismo hipogonadotropo congénito.
Como consecuencia de ellos, los afectados de esa patología genética presentan desarrollo ineficiente de los caracteres sexuales secundarios una vez llegados a la pubertad. Es por ello por lo que los pacientes con síndrome de Kallmann presentan problemas de esterilidad.

Por otra parte, el síndrome de Kallman también causa alteraciones en la percepción del olfato, como la hiposmia y la anosmia, es decir, la disminución o ausencia completa del olfato respectivamente.
¿Cuáles son los genes causantes del síndrome de Kallman?
Hay varias mutaciones genéticas con distintos tipos de herencia que pueden dar lugar al síndrome de Kallman:
- Herencia ligada al sexo: KAL1 (Xp22.32)
- Herencia autosómica dominante: FGFR1 (8p12), FGF8 (10q25-q26), CHD7 (8q12.2) y SOX10 (22q13.1)
- Herencia autosómica recesiva: PROKR2 (20p12.3) y PROK2 (3p21.1)
En función de la mutación y el gen afectado, los portadores del síndrome de Kallman tendrán que plantearse sus opciones reproductivas con tal de evitar que este trastorno se transmita a un hijo.
Lectura recomendada
Como hemos dicho, el síndrome de Kallmann es una enfermedad genética hereditaria. Si quieres conocer todos los detalles sobre los tipos de herencia y el riesgo de transmisión, te animamos a seguir leyendo el siguiente post: ¿Qué enfermedades genéticas o cromosómicas puede detectar el DGP?
Para entender el síndrome de Kallmann, es importante conocer todas las hormonas sexuales que intervienen en el organismo de hombres y mujeres. Puedes encontrar toda esta información aquí: Hormonas sexuales masculinas y femeninas.
Hacemos un gran esfuerzo para ofrecerte información de máxima calidad.
🙏 Por favor, comparte este artículo si te ha gustado. 💜💜 ¡Nos ayudas a seguir!
Bibliografía
Freitas P, Carvalho S, Ribeiro F, Marnoto D, Martins F. Neuroradiology of Kallmann’s syndrome. Acta Med Port 2001;14:123-6 (Ver)
Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Ment Defic 1944;48:203-6 (Ver)
Moorman JR, Crain B, Osborne D. Kallmann’s syndrome with associated cardiovascular and intracranial abnormalities. Am J Med 1984;77: 369-72 (Ver)
Munez A, Dieguez E. A plea for proper recognition: The syndrome of Maestre de San Juan-Kallman. Am J Neuroradiol 1997;18:1395-6 (Ver)
Ping Zhang, Jing-Yun Fu. X-linked recessive Kallmann syndrome: A case report. World J Clin Cases. 2022 Sep 6;10(25):8990-8997 (Ver)
Seminara SB, Hayes FJ, Crowley WF Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. Endocr Rev 1998;19:521–39 (Ver)
Sohyun Moon, Ying-Tao Zhao. Convergent biological pathways underlying the Kallmann syndrome-linked genes Hs6st1 and Fgfr1. Hum Mol Genet. 2022 Dec 16;31(24):4207-4216. doi: 10.1093/hmg/ddac172 (Ver)
Truwit CL, Barkovich AJ, Grumbach MM, Martini JJ. MR imaging of Kallmann Syndrome: A Genetic Disorder of neuronal migration affecting the Olfactory and Genital systems. Am J Neuroradiol 1993;14:827-38 (Ver)